

by Bill Snyder
As cells grow and divide, their DNA needs to be accurately replicated and properly segregated to new cells. Errors during replication or segregation can alter the genome and promote cancer.
Now, researchers at Vanderbilt University Medical Center have identified the mechanism by which the enzyme and tumor suppressor SETD2 prevents the propagation of these errors, and thus safeguards the integrity of the genome.
Their discovery, reported Sept. 18 in the Proceedings of the National Academy of Sciences (PNAS), provides a remarkably detailed view of the earliest events leading to the development of cancer, and of potential new ways to prevent it.
The researchers found that loss of SETD2 during DNA replication was sufficient to promote the formation of abnormal, mirror-imaged chromosomes called isochromosomes. Although isochromosomes are commonly observed in cancers of the blood and in solid tumors, as well as in developmental syndromes, it was previously not known how they formed.
“This is one of the first studies to uncover a mechanism responsible for their formation in mammalian cells, and the first linking it to a gene relevant in cancer,” said the paper’s first author, Frank Mason, PhD, assistant professor of Medicine at VUMC, and corresponding author for this study.
With Kimryn Rathmell, MD, PhD, the Hugh Jackson Morgan Professor of Medicine and chair of the Department of Medicine, Mason has been studying SETD2 since his arrival at VUMC in 2016.
In 2018, they and their colleagues reported that an “insufficiency” of SETD2 in kidney cells led to chromosome segregation errors and was an early driver of genomic instability in clear cell renal cell carcinoma, the most common form of kidney cancer.
However, the mechanism linking the mutation or deletion of the SETD2 gene to mitotic errors during cell division and resulting chromosomal abnormalities remained unknown—until now.
Normally SETD2 places an “epigenetic” mark on DNA by methylating, or chemically modifying, histones, the protein “spools” around which the DNA “string” wraps to prevent it from becoming entangled during replication.
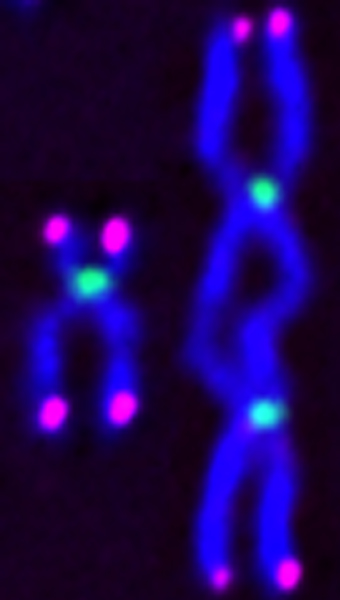
In the current study, the researchers found that loss of SETD2 or its mark led to the formation of abnormal chromosomal bridges at the completion of cell division. Further exploration of the chromosome structure revealed an unusual type of chromosomal error with mirror-imaged or inverted symmetry—the isochromosome.
Isochromosomes can occur when DNA replication takes an unauthorized U-turn, creating daughter chromosomes that duplicate the genetic material of one arm on both sides of the centromere, the constricted region of the chromosome which plays a key role in segregating the DNA during cell division.
This leads to a duplication of genes, but more importantly, the half of the chromosome that was not replicated is lost, contributing to gene deletions and aneuploidy, missing chromosomes.
When the U-turn happens to duplicate the centromere, daughter chromosomes are produced that have two centromeres, known as dicentric chromosomes. These structures are prone to breakage, as they can be pulled apart by opposite spindle poles during mitosis, the replication of chromosomes into two new nuclei.
Finding rearranged isochromosomes with mirror-imaged or inverted symmetry in SETD2-deficient cells, the researchers showed that the cells had altered chromosome and gene numbers and, furthermore, that the isochromosomes were responsible for the mis-segregated chromosomes during cell division.
Together, these findings reveal critical new insights into how the genome is normally protected from these kinds of errors.
“It took a strong cell biologist like Dr. Mason to recognize and visualize this unusual event in chromosomes,” Rathmell said. “But once you see one, especially the dicentric chromosome with two centromeres, it is so striking, you want to look for more.
“His work blending molecular techniques with cell biology approaches led to the discovery that single centromere isochromosomes, which are harder to recognize using standard techniques, are an even more common event in SETD2 mutant tumors,” she said.
The diversity in chromosome number and rearrangements observed in isochromosomes generates heterogeneity within cell populations and provides tumors with a “fitness advantage” over normal cells. Accordingly, a range of tumor types with SETD2 mutations are highly resistant to a broad range of cancer drugs.
Notably, clear cell renal cell carcinoma, the predominant form of the disease in which SETD2 was initially found to be a tumor suppressor, is among the cancers that are most resistant to chemotherapy.
The authors propose that isochromosomes may be a critical mechanism promoting chemotherapeutic resistance, and they are devising ways to target these pathways.
In the future, this new understanding of the underlying tumor biology may help physicians select the most effective therapy for their patients with cancer and devise ways to reverse the resistance to cancer chemotherapy.
Co-authors of the paper from VUMC are Courtney Lovejoy, PhD, Emily Kounlavong, Anteneh Tebeje, Tiffany Guess, PhD, Logan Vlach, and Stephen Norris, PhD.
Funding for this project was provided by the Kidney Cancer Association, the Cancer Prevention and Research Institute of Texas, the U.S. Department of Defense, and National Institutes of Health grants 5T32 CA009592, R01 CA275082, and R01 CA020301.

